1 Name of Medicine
Onasemnogene abeparvovec.
2 Qualitative and Quantitative Composition
Onasemnogene abeparvovec is a gene therapy medicinal product that expresses the human survival motor neuron (SMN) protein. It is a non-replicating recombinant adeno-associated vector serotype 9 (AAV9) containing the cDNA of the human SMN gene under the control of the cytomegalovirus enhancer/chicken-β-actin-hybrid promoter.
Onasemnogene abeparvovec is produced in human embryonic kidney cells by recombinant DNA technology.
Each mL contains onasemnogene abeparvovec with a nominal concentration of 2 x 1013 vector genomes (vg). Vials will contain an extractable volume of not less than either 5.5 mL or 8.3 mL. The total number of vials and combination of fill volumes in each finished pack will be customised to meet dosing requirements for individual patients depending on their weight (see Section 4.2 Dose and Method of Administration; Section 6.5 Nature and Contents of Container).
Excipient with known effect.
This medicinal product contains 0.2 mmol sodium per mL.
For the full list of the excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Injection for intravenous infusion.
When thawed, it is a clear to slightly opaque, colourless to faint white solution.
4.1 Therapeutic Indications
Zolgensma (onasemnogene abeparvovec) is indicated for the treatment of paediatric patients less than 9 months of age with symptomatic or pre-symptomatic spinal muscular atrophy with bi-allelic mutations in the survival motor neuron 1 (SMN1) gene and 1 to 3 copies of the SMN2 gene.
4.2 Dose and Method of Administration
Dose.
For single-dose intravenous infusion only.
Treatment with Zolgensma should be supervised by a physician experienced in the management of patients with SMA.
In order to improve the traceability of biological medicinal products, the name and the batch number of the administered product should be clearly recorded.
An immune response to the adeno-associated viral vector serotype 9 (AAV9) capsid will occur after infusion of Zolgensma, thus patients should not be re-dosed with Zolgensma.
Zolgensma is for a single treatment only.
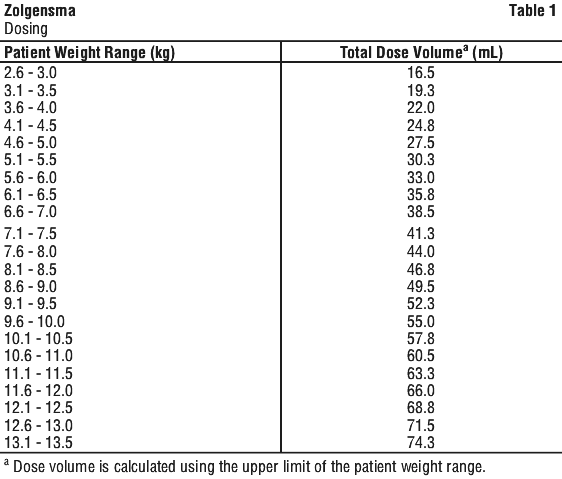
The recommended dose of Zolgensma is 1.1 x 1014 vector genomes per kilogram (vg/kg) of body weight (see Table 1).
 Due to the increased risk of serious systemic immune response, it is recommended that patients are clinically stable in their overall health status (e.g. hydration and nutritional status, absence of infection) prior to Zolgensma infusion. Zolgensma should be postponed in patients with infections until the infection has resolved and the patient is clinically stable. Clinical signs or symptoms of infection should not be evident at the time of Zolgensma infusion (see Section 4.4 Special Warnings and Precautions for Use, Systemic immune response).
Due to the increased risk of serious systemic immune response, it is recommended that patients are clinically stable in their overall health status (e.g. hydration and nutritional status, absence of infection) prior to Zolgensma infusion. Zolgensma should be postponed in patients with infections until the infection has resolved and the patient is clinically stable. Clinical signs or symptoms of infection should not be evident at the time of Zolgensma infusion (see Section 4.4 Special Warnings and Precautions for Use, Systemic immune response).
Patients with ALT, AST or total bilirubin greater than 2 times ULN should not be dosed with Zolgensma. The exception is when the elevated total bilirubin is due to neonatal jaundice.
Laboratory testing and monitoring to assess safety.
Prior to Zolgensma infusion, the following tests should be conducted at baseline (see Section 4.4 Special Warnings and Precautions for Use, Hepatotoxicity, Immunogenicity, Thrombocytopenia, Thrombotic microangiopathy, Elevated troponin-I):
AAV9 antibody testing (retesting may be performed if AAV9 antibody titres are reported as > 1:50).
Liver function (clinical exam, aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, prothrombin time, albumin, partial thromboplastin time (PTT) and international normalised ratio (INR)).
Creatinine.
Complete blood count (including haemoglobin and platelet count).
Troponin-I.
After Zolgensma infusion the following tests should be conducted on a regular basis (see Section 4.4 Special Warnings and Precautions for Use, Hepatotoxicity, Thrombocytopenia, Elevated troponin-I).
Liver function (clinical exam, aspartate aminotransferase (AST), alanine aminotransferase (ALT), total bilirubin, prothrombin time) weekly for the month after Zolgensma infusion and then weekly during the corticosteroid taper period. If the patient is clinically stable with unremarkable findings at the end of the corticosteroid taper period, liver function should continue to be monitored every two weeks for another month.
Platelet counts weekly for the first month, and then every other week for the second and third months, until platelet counts return to baseline.
Troponin-I weekly for the first month, and then monthly for the second and third months, until troponin-I level returns to baseline.
Systemic corticosteroid treatment pre- and post-Zolgensma infusion.
Transient elevations in liver aminotransferases after treatment with Zolgensma were experienced by some patients (see Section 4.8 Adverse Effects (Undesirable Effects)). To manage a possible increase in liver aminotransferases, all patients should receive systemic corticosteroids given orally before and after dosing Zolgensma (see Section 4.4 Special Warnings and Precautions for Use, Hepatotoxicity).
Treatment with systemic corticosteroid prior to Zolgensma infusion.
One day prior to Zolgensma infusion, pre-treat with oral prednisolone at a dose of 1 mg/kg/day (or equivalent if another corticosteroid is used).
Continued treatment with systemic corticosteroid and liver function monitoring following Zolgensma infusion.
Prednisolone should be administered daily at 1 mg/kg/day (or equivalent if another corticosteroid is used) for 30 days following infusion with Zolgensma.
At the end of the 30-day period of systemic corticosteroid treatment, check liver status clinically and by assessing ALT, AST, total bilirubin, and prothrombin time. Liver function should be monitored for at least 3 months following Zolgensma infusion, and at other times as clinically indicated (see Section 4.4 Special Warnings and Precautions for Use, Hepatotoxicity).
Promptly clinically assess and closely monitor patients with worsening liver function test results and/or signs or symptoms of acute illness.
For patients with unremarkable findings (normal clinical exam, total bilirubin, and prothrombin time, and ALT and AST levels below 2 x ULN): Taper the prednisolone (or equivalent if another corticosteroid is used) dose over the next 28 days, e.g. 2 weeks at 0.5 mg/kg/day and then 2 weeks at 0.25 mg/kg/day oral prednisolone. Systemic corticosteroids should not be stopped abruptly, rather tapered gradually (see Section 4.4 Special Warnings and Precautions for Use, Hepatotoxicity).
If liver function abnormalities persist, continue systemic corticosteroids (equivalent to oral prednisolone at 1 mg/kg/day) until AST and ALT values are both below 2 x ULN and all other assessments return to normal range, and then taper the corticosteroid dose over the next 28 days or longer if needed. Systemic corticosteroids should not be stopped abruptly, rather tapered gradually.
Promptly consult a paediatric gastroenterologist or hepatologist if patients do not respond adequately to the equivalent of 1 mg/kg/day oral prednisolone. If oral corticosteroid therapy is not tolerated or not effective, intravenous corticosteroids may be considered as clinically indicated.
Variance from these recommendations is at the discretion of the treating physician. If another corticosteroid is used by the physician in place of prednisolone, similar considerations and approach to taper the corticosteroid dose after 30 days following infusion with Zolgensma should be taken as appropriate.
Where feasible, the patient's vaccination schedule should be adjusted to accommodate concomitant corticosteroid administration prior to and following Zolgensma infusion (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Requirements for a genetically-modified organism.
Zolgensma contains a genetically-modified organism. Follow local institutional guidelines for storage, handling, administration and disposal of medicinal products containing genetically-modified organisms.
The onasemnogene abeparvovec syringe should be prepared aseptically under sterile conditions.
Personal protective equipment (to include gloves, safety goggles, laboratory coat and sleeves) should be worn while preparing or administering onasemnogene abeparvovec. Personnel should not work with onasemnogene abeparvovec if skin is cut or scratched.
All spills of onasemnogene abeparvovec must be wiped with absorbent gauze pad and the spill area must be disinfected using a bleach solution followed by alcohol wipes. All clean up materials must be double bagged and disposed of per institutional guidelines for biohazard waste.
All materials that may have come in contact with onasemnogene abeparvovec (e.g. vial, all materials used for injection, including sterile drapes and needles) must be disposed of in accordance with local institutional biosafety guidelines.
Take precautions to avoid accidental exposure to onasemnogene abeparvovec. In the event of exposure to skin, the affected area must be thoroughly cleaned with soap and water for at least 15 minutes. In the event of exposure to eyes, the affected area must be thoroughly flushed with water for at least 15 minutes.
Method of administration.
Administer Zolgensma as a single-dose intravenous infusion through a venous catheter.
Zolgensma is for single use in one patient only. Discard any residue.
Preparation.
Zolgensma should be prepared aseptically under sterile conditions.
Thaw Zolgensma before use. The contents of the Zolgensma pack will thaw in approximately 12 hours when placed in a refrigerator (2°C - 8°C), or in approximately 4 hours if placed at room temperature (20°C - 25°C).
If thawed in a refrigerator, remove from refrigerator on day of dosing.
When thawed, Zolgensma is a clear to slightly opaque, colourless to faint white liquid, free of particles. Visually inspect vials for particulate matter and discolouration prior to infusion. Do not use vials if particulates or discolouration are present.
Do not shake.
Prior to dosing draw the appropriate dose volume from all vials into a syringe, remove air from the syringe, cap the syringe, and deliver the syringe at room temperature to the patient infusion location. Dispose of vials in accordance with local institutional biosafety guidelines.
Use Zolgensma within 6 hours of drawing into syringe. Discard the vector-containing syringe if the drug is not infused within the 6 hour timeframe. Dispose of the syringe in accordance with local institutional biosafety guidelines.
Do not refreeze.
Follow the steps below for infusion.
1. Place a primary catheter into a vein (generally a peripheral vein in the arm or leg). Insertion of a back-up catheter is recommended.
2. Program syringe pump for saline priming, or prime tubing manually with saline.
3. Administer Zolgensma as a slow infusion over 60 minutes. Do not infuse as an intravenous push or bolus.
4. Flush line with saline following completion of infusion.
Special populations.
Paediatric patients.
Administration of Zolgensma to premature neonates before reaching full-term gestational age should be carefully considered. The safety and efficacy of Zolgensma in these patients have not been established.
The efficacy and safety of Zolgensma was studied in paediatric patients who received Zolgensma infusion at age 0.3 to 7.9 months (weight up to 8.4 kg) (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties, Clinical trials).4.3 Contraindications
Hypersensitivity to the active substance or to any of the excipients listed in Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Advanced SMA.
Since SMA results in progressive and non-reversible damage to motor neurons, the benefit of Zolgensma in symptomatic patients depends on the degree of disease burden at the time of treatment, with earlier treatment resulting in potential higher benefit.
Progressive motor neuron loss is irreversible. The treating physician should consider that the benefit is seriously reduced in patients with profound muscle weakness and respiratory failure, patients on permanent ventilation, and patients not able to swallow.
Patients requiring permanent ventilatory support have not been evaluated and treatment with Zolgensma is not recommended. Permanent ventilation is defined as requiring invasive ventilation (tracheostomy), or respiratory assistance for 16 or more hours per day (including noninvasive ventilatory support) continuously for 14 or more days in the absence of an acute reversible illness, excluding perioperative ventilation.
Hepatotoxicity.
Administration of AAV vector may result in aminotransferase elevations, which may be serious. In clinical trials, elevated transaminases greater than 2 times ULN were observed in 31% of patients treated at the recommended dose (see Section 4.8 Adverse Effects (Undesirable Effects)).
Acute serious liver injury and acute liver failure have occurred with Zolgensma use. Cases of acute liver failure with fatal outcomes have been reported.
Patients with pre-existing hepatic impairment or acute hepatic viral infection may be at higher risk of acute serious liver injury/acute liver failure.
Patients with ALT, AST or total bilirubin greater than 2 times ULN should not be dosed with Zolgensma. The exception is when the elevated total bilirubin is due to neonatal jaundice.
Prior to infusion, liver function of all patients should be assessed by clinical examination and laboratory testing (AST, ALT, total bilirubin, prothrombin time, albumin, PTT and INR).
In order to mitigate potential aminotransferase elevations, a systemic corticosteroid should be administered to all patients before and after Zolgensma infusion (see Section 4.2 Dose and Method of Administration).
Liver function (clinical exam, ALT, AST, total bilirubin and prothrombin time) should be monitored for at least 3 months after infusion and at other times as clinically indicated (see Section 4.2 Dose and Method of Administration).
Promptly clinically assess and closely monitor patients with worsening liver function test results and/or signs or symptoms of acute illness.
In case hepatic injury is suspected, further testing is recommended (e.g. albumin, PTT and INR).
The risks and benefits of Zolgensma therapy should be carefully considered in patients with pre-existing hepatic impairment.
Immune-mediated hepatotoxicity that generally manifested as elevated ALT and/or AST levels has been reported with Zolgensma use. Immune-mediated hepatotoxicity may require adjustment of the corticosteroid treatment regimen including longer duration, increased dose or prolongation of the corticosteroid taper (see Section 4.2 Dose and Method of Administration; Section 4.8 Adverse Effects (Undesirable Effects)).
Laboratory tests AST, ALT, total bilirubin, albumin, prothrombin time, PTT and INR should be assessed before Zolgensma infusion. AST, ALT, prothrombin time and total bilirubin should be monitored weekly for the month after Zolgensma infusion and then weekly during the corticosteroid taper period. If the patient is clinically stable with unremarkable findings at the end of the corticosteroid taper period, liver function should continue to be monitored every two weeks for another month. Tapering of systemic corticosteroids should not be considered until AST/ALT levels are less than 2 x ULN and all other assessments are unremarkable (see Section 4.2 Dose and Method of Administration, Systemic corticosteroid treatment pre- and post-Zolgensma infusion).
Systemic immune response.
Due to the increased risk of serious systemic immune response, it is recommended that patients are clinically stable in their overall health status (e.g. hydration and nutritional status, absence of infection) prior to Zolgensma infusion. Zolgensma should be postponed in patients with infections until the infection has resolved and the patient is clinically stable. Clinical signs or symptoms of infection should not be evident at the time of Zolgensma infusion (see Section 4.2 Dose and Method of Administration).
Infection, either acute (e.g. respiratory) or chronic uncontrolled, could increase the risk of serious systemic immune response, potentially resulting in more severe clinical courses of the infection. Patients with infection were excluded from participation in Zolgensma clinical trials. Increased vigilance in the prevention, monitoring and management of infection is recommended before and after Zolgensma infusion. Seasonal prophylaxis against respiratory syncytial virus (RSV) is recommended and should be up-to-date.
The treating physician should be aware of the possibility of adrenal insufficiency related to longer duration of treatment with corticosteroids or increased dose.
Immunogenicity.
In Zolgensma clinical trials, confirmation of AAV9 antibody titres at or below 1:50 was required prior to infusion. It has not been established whether infusion of Zolgensma may represent a risk for an immune response for patients with pre-existing AAV9 antibodies at higher titres. The safety and efficacy of Zolgensma has not been established in patients with baseline AAV9-antibody titres above 1:50. Patients should be tested for the presence of AAV9 antibodies prior to infusion with Zolgensma. Retesting may be performed if AAV9 antibody titres are reported as above 1:50. An immune response to the adeno-associated viral vector serotype 9 (AAV9) capsid will occur after infusion of Zolgensma.
Thrombocytopenia.
Transient decreases in platelet counts, some of which met the criteria for thrombocytopenia, were typically observed within the first two weeks after Zolgensma infusion (see Section 4.8 Adverse Effects (Undesirable Effects)).
Platelet counts should be obtained before Zolgensma infusion and should be closely monitored for significant decreases within the first two weeks following infusion and on a regular basis afterwards; at least weekly for the first month and every other week for the second and third months until platelet counts return to baseline (see Section 4.2 Dose and Method of Administration, Laboratory testing and monitoring to assess safety).
Thrombotic microangiopathy.
Cases of thrombotic microangiopathy (TMA) have been reported to occur generally within the first two weeks after Zolgensma infusion in the post-marketing setting (see Section 4.8 Adverse Effects (Undesirable Effects)). Thrombotic microangiopathy is characterized by thrombocytopenia, microangiopathic haemolytic anaemia, and acute kidney injury.
Concurrent immune system activation (e.g. infections, vaccinations) was identified as a contributing factor in some cases.
Prompt attention to signs and symptoms of TMA is advised, as TMA can result in life-threatening or fatal outcomes.
Thrombocytopenia is a key feature of TMA, therefore platelet counts should be closely monitored for significant decreases within the first two weeks following infusion and on a regular basis afterwards (see Section 4.4, Thrombocytopenia above), as well as signs and symptoms of TMA, such as hypertension, increased bruising, seizures or decreased urine output. In case these signs and symptoms occur in the presence of thrombocytopenia, further diagnostic evaluation for haemolytic anaemia and renal dysfunction should be promptly undertaken. If clinical signs, symptoms and/or laboratory findings consistent with TMA occur, a paediatric haematologist and/or paediatric nephrologist should be consulted immediately to manage TMA as clinically indicated.
Elevated troponin-I.
Increases in cardiac troponin-I levels (up to 0.2 microgram/L) were observed following Zolgensma infusion in clinical trials (see Section 4.8 Adverse Effects (Undesirable Effects)). Elevated troponin-I levels found in some patients may indicate potential myocardial tissue injury. Clinical cardiac findings of concern have not been observed following administration of Zolgensma. Cardiac toxicity was observed in animal studies (see Section 5.3 Preclinical Safety Data). Monitor troponin-I before Zolgensma infusion and on a regular basis for at least 3 months afterwards (weekly for the first month, and then monthly for the second and third months until troponin-I level returns to baseline), see Section 4.2 Dose and Method of Administration, Laboratory testing and monitoring to assess safety. Consider consultation with a cardiac expert as needed.
Theoretical risk of tumorigenicity as a result of vector integration.
There is a theoretical risk of tumorigenicity due to integration of AAV vector DNA into the genome.
Zolgensma is composed of a non-replicating AAV9 vector whose DNA persists largely in episomal form. Rare instances of random vector integration into human DNA are possible with recombinant AAV. The clinical relevance of individual integration events is unknown, but it is acknowledged that individual integration events could potentially contribute to a risk of tumorigenicity.
Systemic corticosteroid administration and live vaccines.
Live vaccines should not be administered to patients receiving high doses of corticosteroids (i.e. ≥ 2 weeks of daily receipt of 20 mg or 2 mg/kg body weight of prednisone or equivalent) prior to and following Zolgensma infusion (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Use in hepatic impairment.
Zolgensma has not been studied in patients with hepatic impairment. Zolgensma therapy should be carefully considered in patients with hepatic impairment. One patient who received Zolgensma developed acute serious liver injury; that patient had elevated aminotransferase levels prior to Zolgensma infusion. In clinical trials, elevation of aminotransferases was observed in patients following Zolgensma infusion (see Section 4.4, Hepatotoxicity).
Use in renal impairment.
The safety and efficacy of Zolgensma have not been established in patients with renal impairment.
Use in the elderly.
Not applicable to this medicine.
Paediatric use.
Administration of Zolgensma to premature neonates before reaching full-term gestational age is not recommended because concomitant treatment with corticosteroids may adversely affect neurological development. Delay Zolgensma infusion until the corresponding full-term gestational age is reached.
There is no information on whether breastfeeding should be restricted in mothers who may be seropositive for anti-AAV9 antibodies.
The efficacy and safety of Zolgensma was studied in paediatric patients who received Zolgensma infusion at age 0.3 to 7.9 months (weight up to 8.4 kg) (see Section 4.8 Adverse Effects (Undesirable Effects); Section 5.1 Pharmacodynamic Properties, Clinical trials).
Effects on laboratory tests.
No data are available.4.5 Interactions with Other Medicines and Other Forms of Interactions
No interaction studies have been performed. Interactions with antiviral medicinal products are not expected. Live vaccines, such as MMR and varicella, are contraindicated for patients on a substantially immunosuppressive steroid dose (i.e. ≥ 2 weeks of daily receipt of 20 mg or 2 mg/kg body weight of prednisone or equivalent) because high doses of corticosteroids may reduce the immune response to these vaccines. Where feasible, adjust a patient's vaccination schedule to accommodate concomitant corticosteroid administration prior to and following Zolgensma infusion (see Section 4.2 Dose and Method of Administration). Seasonal RSV prophylaxis is not precluded.
Zolgensma has not been studied in conjunction with other therapies for SMA. No information is available whether use of multiple therapies might carry additional risks.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no fertility data available.
(Category B2)
There are no available data regarding Zolgensma use in pregnant women. No animal reproductive and developmental toxicity studies have been conducted with Zolgensma.
There is no information available on the presence of Zolgensma in human milk, the effects on the breastfed infant, or the effects on milk production.4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
The safety of Zolgensma was evaluated in 99 patients who received the recommended dose (1.1 x 1014 vg/kg) from 5 open-label clinical studies (CL-101, CL-303, CL-302, CL-304, CL-306). The patients ranged in age from 0.3 months to 7.9 months at the time of administration (weight range: 3.0 kg to 8.4 kg). Through the cutoff date of 11-Jun-2020, patients receiving the therapeutic dose had been followed for a median of 14.6 months, with follow-up ranging from 1.0 month in Study CL-306 to 25.7 months in Study CL-101.
Of the 99 patients who received the therapeutic dose, 46 patients (46.5%) had at least 1 SAE, and 39 patients (39.4%) had at least one Treatment-Emergent Adverse Event (TEAE) that was Grade 3 or 4. See Table 3 for the most frequently reported TEAEs (≥ 5%) at the proposed therapeutic IV dose.
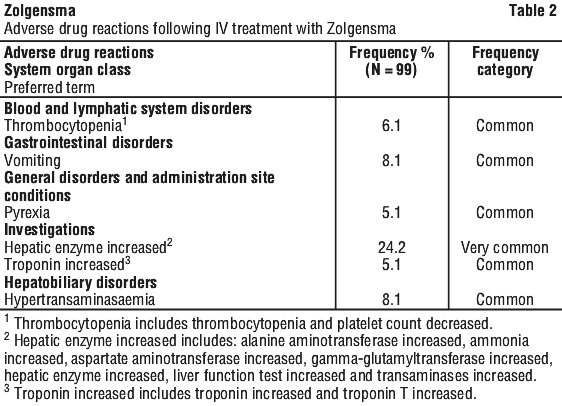
The adverse drug reactions identified with Zolgensma in patients treated with intravenous infusion at the recommended dose from the 5 open-label clinical studies are presented in Table 2. Among these 99 patients, the most frequently reported (≥ 5%) adverse reactions following administration were hepatic enzyme increased, hypertransaminasaemia, vomiting, thrombocytopenia, troponin increased and pyrexia.
Adverse drug reactions from clinical trials are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness. Frequency categories are derived according to the following conventions (CIOMS III): very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000).

Adverse drug reactions from post-marketing (frequency not known).
The following adverse drug reactions have been derived from post-marketing experience with Zolgensma including spontaneous case reports and literature cases. Because these reactions are reported voluntarily, it is not possible to reliably estimate their frequency which is therefore categorised as not known. The adverse drug reactions are listed according to system organ classes in MedDRA. Within each system organ class, ADRs are presented in order of decreasing seriousness. See Table 4.

Description of selected adverse drug reactions.
Hepatobiliary disorders.
In clinical trials, elevated transaminases greater than 2 times ULN were observed in 31% of patients treated at the recommended dose.
Some patients have experienced AST and ALT elevations > 20 x ULN and have been symptomatic (e.g. vomiting, jaundice), which required the use of corticosteroids, sometimes with prolonged duration and/or a higher dose.
Outside of clinical trials, including in the post marketing setting, there have been reports of children developing signs and symptoms of acute liver failure (e.g. jaundice, coagulopathy, encephalopathy) typically within 2 months of treatment with Zolgensma, despite receiving prophylactic corticosteroids before and after infusion. Cases of acute liver failure with fatal outcomes have been reported.
Transient thrombocytopenia.
In clinical trials, transient decreases from baseline in mean platelet counts (some of which met the criteria for thrombocytopenia) that reached levels of potential clinical significance were seen in 5.1% of patients at multiple time points post-dose and normally resolved within two weeks; the decreases were reported as thrombocytopenia in 6.1% of patients. Decreases in platelet counts were more prominent during the first week of treatment (see Section 4.4 Special Warnings and Precautions for Use).
Increases in troponin-I levels.
Increases in cardiac troponin-I levels up to 0.2 microgram/L following Zolgensma infusion were observed. The clinical relevance of these findings is not known (see Section 4.4 Special Warnings and Precautions for Use).
Immunogenicity.
The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. In addition, the observed incidence of antibody (including neutralising antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medicinal products and underlying disease. Pre- and post-gene therapy titres of AAV9 antibody were measured in the clinical studies (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties, Clinical trials).
In Zolgensma clinical trials, patients were required to have baseline anti-AAV9 antibody titres of ≤ 1:50, measured using an enzyme-linked immunosorbent assay (ELISA). Evidence of prior exposure to AAV9 was uncommon. Mean increases from baseline in anti-AAV9 antibody titres were observed in all patients at all but one time point for antibody titre levels to AAV9 peptide, reflecting normal response to non-self viral antigen. Some patients experienced anti-AAV9 titres exceeding the level of quantification, however most of these patients did not have potentially clinically significant adverse drug reactions. Thus, no relationship has been established between high AAV9 antibody titres and the potential for adverse drug reactions or efficacy parameters.
In the CL-101 clinical study, 16 patients were screened for AAV9 antibody titre: 13 had titres less than 1:50 and were enrolled in the study; three patients had titres greater than 1:50, two of whom were retested following cessation of breast-feeding and their titres were measured at less than 1:50 and both were enrolled in the study. There is no information on whether breastfeeding should be restricted in mothers who may be seropositive for AAV9 antibodies. Patients all had less than or equal to 1:50 AAV9 antibody titre prior to treatment with Zolgensma and subsequently demonstrated an increase in AAV9 antibody titres to at least 1:102,400 and up to greater than 1:819,200. No Zolgensma-treated patient demonstrated an immune response to the transgene. Re administration of Zolgensma in the presence of high anti-AAV9 antibody titre has not been evaluated.
Other special populations.
Body weight ≥ 8.5 kg to ≤ 21 kg.
The safety of Zolgensma was evaluated in a post-authorisation clinical study (COAV101A12306) in 24 patients weighing ≥ 8.5 kg to ≤ 21 kg (median weight: 15.8 kg). The patients ranged in age from approximately 1.5 to 9 years at the time of administration. 1 of the 24 patients was under the age of 2 at the time of administration (median age: 4.9 years). Patients had 2 to 4 copies of SMN2. Before treatment with Zolgensma, 21 patients discontinued their previous treatment with nusinersen or risdiplam. The types of adverse reactions observed were consistent with that of the 5 open-label studies.
AST or ALT elevations > 2 x ULN were observed in the majority of patients (23/24). These patients were clinically asymptomatic and there were no elevations of bilirubin. The AST and ALT elevations were managed with the use of corticosteroids, typically with prolonged duration and/or a higher dose (see Section 4.4 Special Warnings and Precautions for Use).
Transient decreases in platelet counts, which met the criteria for thrombocytopenia were observed in 20 out of 24 patients (see Section 4.8 Adverse Effects (Undesirable Effects), Transient thrombocytopenia).
Reporting of suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at https://www.tga.gov.au/reporting-problems.4.9 Overdose
No data from clinical studies are available regarding overdose of Zolgensma. The dose of the medicinal product is specific to each individual patient's weight and is administered once only (see Section 4.2 Dose and Method of Administration), therefore overdose is considered unlikely. Adjustment of the dose of prednisolone, close clinical observation and monitoring of laboratory parameters (including clinical chemistry and haematology) for systemic immune response are recommended (see Section 4.4 Special Warnings and Precautions for Use).
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Other drugs for disorders of the musculo-skeletal system, ATC code: M09AX09.
Mechanism of action.
SMA is caused by a bi-allelic mutation in the SMN1 gene, which results in insufficient SMN protein expression. Onasemnogene abeparvovec is a non-replicating recombinant AAV vector that utilises the AAV9 capsid to deliver a stable fully functional copy of the transgene encoding the human survival motor gene (SMN1) protein. The SMN1 gene present in onasemnogene abeparvovec is designed to reside as episomal DNA in the nucleus of transduced cells and is expected to be stably expressed for an extended period of time in post-mitotic cells. Rare instances of random vector integration into human DNA are possible with recombinant AAV (see Section 4.4 Special Warnings and Precautions for Use). The AAV9 virus is not known to cause disease in humans. Intravenous administration of Zolgensma that results in cell transduction and expression of the SMN protein has been observed in two human case studies (see Section 5.2 Pharmacokinetic Properties).
Pharmacodynamics (PD).
There are no clinically relevant pharmacodynamics data for onasemnogene abeparvovec.
Clinical trials.
Zolgensma has not been studied in patients with a bi-allelic mutation of the SMN1 gene and only 1 copy of SMN2 or ≥ 4 copies SMN2 in clinical trials.
AVXS-101-CL-303 phase 3 study in patients with type 1 SMA.
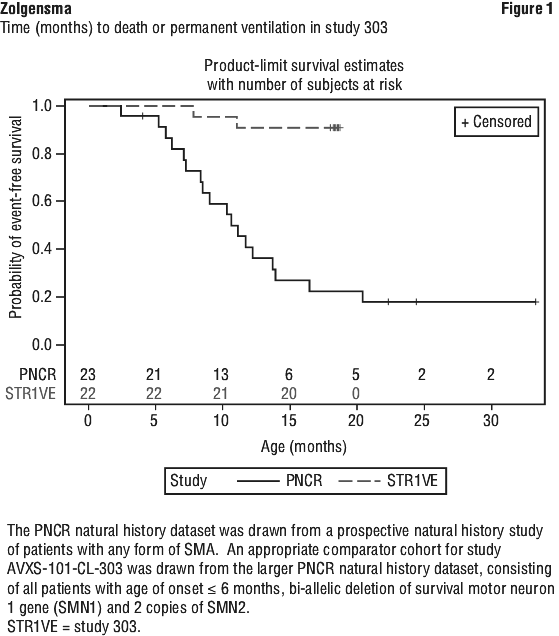
AVXS-101-CL-303 (Study CL-303) is a phase 3, open-label, single-arm, single-dose study of intravenous administration of Zolgensma at the therapeutic dose (1.1 x 1014 vg/kg). All patients had genetically confirmed bi-allelic SMN1 gene deletions, 2 copies of the SMN2 gene, absence of the c.859G > C modification in exon 7 of the SMN2 gene and onset of clinical symptoms consistent with SMA prior to 6 months of age. Patient ages and weight at administration ranged from 0.5 to 5.9 months and 3.9 to 7.5 kg. Of the 22 enrolled patients, three patients discontinued the study and two patients had an event (death or permanent ventilation) leading to 90.9% (95% CI: 79.7%, 100.0%) event-free survival (alive without permanent ventilation) at 14 months of age (see Figure 1). This is markedly improved compared to the natural history of infantile onset SMA as reported in the Pediatric Neuromuscular Clinical Research natural history cohort (PNCR), in which only 25% of subjects were alive without permanent ventilation by the age of 14 months.
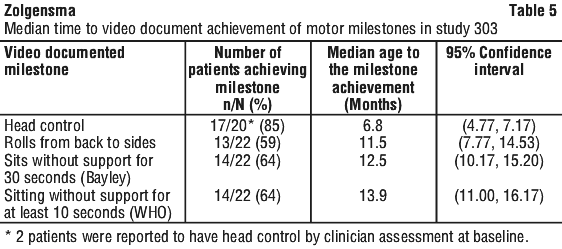
 For the 14 patients in Study CL-303 that achieved the milestone of independent sitting for at least 30 seconds, the median age when this milestone was first demonstrated was 12.5 months (range 9.2 to 18.6 months). Thirteen patients confirmed the milestone of independent sitting for at least 30 seconds at the 18 month visit (co-primary endpoint, p < 0.0001). One patient achieved the milestone of sitting independently for 30 seconds at 16 months of age, but this milestone was not confirmed at the Month 18 visit. The video-confirmed developmental milestones for patients in Study CL-303 are summarised in Table 5.
For the 14 patients in Study CL-303 that achieved the milestone of independent sitting for at least 30 seconds, the median age when this milestone was first demonstrated was 12.5 months (range 9.2 to 18.6 months). Thirteen patients confirmed the milestone of independent sitting for at least 30 seconds at the 18 month visit (co-primary endpoint, p < 0.0001). One patient achieved the milestone of sitting independently for 30 seconds at 16 months of age, but this milestone was not confirmed at the Month 18 visit. The video-confirmed developmental milestones for patients in Study CL-303 are summarised in Table 5.
 One patient (4.5%) could also walk with assistance at 12.9 months. Based on the natural history of the disease, patients who met the study entry criteria would not be expected to attain the ability to sit without support, and only approximately 25% of these patients would be expected to survive (i.e. being alive without permanent ventilation) beyond 14 months of age.
One patient (4.5%) could also walk with assistance at 12.9 months. Based on the natural history of the disease, patients who met the study entry criteria would not be expected to attain the ability to sit without support, and only approximately 25% of these patients would be expected to survive (i.e. being alive without permanent ventilation) beyond 14 months of age.
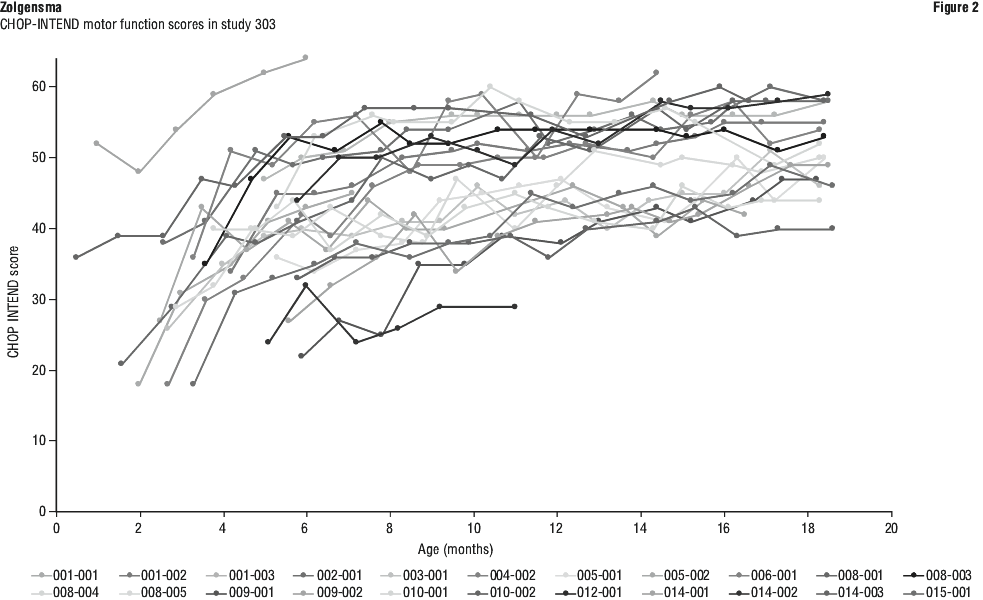
Motor function improvements were also observed as measured by the Children's Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND), see Figure 2. Twenty-one patients (95.5%) achieved a CHOP-INTEND score ≥ 40, 14 (64%) had achieved a CHOP-INTEND score ≥ 50, and 5 patients (23%) had achieved a CHOP-INTEND score ≥ 60. Patients with untreated SMA Type 1 almost never achieve a CHOP-INTEND score ≥ 40.
AVXS-101-CL-101 phase 1 study in patients with type 1 SMA.
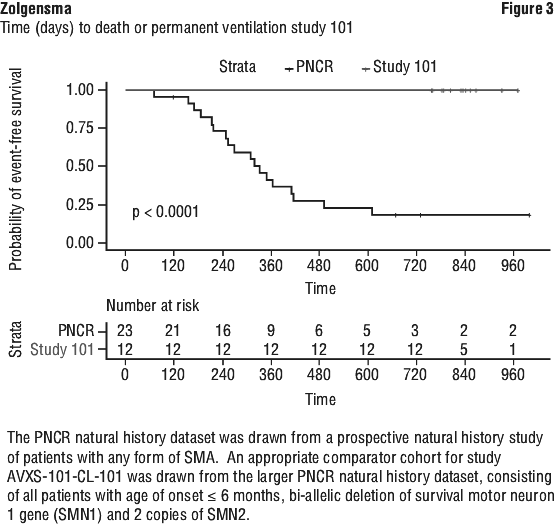
The results seen in Study CL-303 are supported by study AVXS-101-CL-101. All patients had genetically confirmed bi-allelic SMN1 gene deletions, 2 copies of the SMN2 gene, absence of the c.859G > C modification in exon 7 of the SMN2 gene and onset of clinical symptoms consistent with SMA prior to 6 months of age. Zolgensma was administered in study AVXS-101-CL-101 (Phase 1 study in Type 1 SMA, Study CL-101) as a single intravenous infusion in 12 patients from 3.6 kg to 8.4 kg (0.9 to 7.9 months of age). Patients were followed for 24 months. At 14 months of age, all treated patients were event free; i.e. survived without permanent ventilation, compared to 25% in the PNCR natural history cohort. At the end of the study (24 months post dose), all treated patients were event free, compared to less than 8% in the natural history cohort (see Figure 3).
 At 24 months of follow up post dose, 10 patients treated with the therapeutic dose were able to sit without support for ≥ 10 seconds, 9 patients were able to sit without support for ≥ 30 seconds and 2 patients were able to stand alone, walk with assistance and walk alone.
At 24 months of follow up post dose, 10 patients treated with the therapeutic dose were able to sit without support for ≥ 10 seconds, 9 patients were able to sit without support for ≥ 30 seconds and 2 patients were able to stand alone, walk with assistance and walk alone.
Ten of 12 patients from Study CL-101 who received the proposed therapeutic dose of Zolgensma continue to be followed in a long-term study (for up to 5.7 years after dosing) and all have either maintained all previously attained milestones or even gained new milestones including sitting with support, stand with assistance and walk alone.
AVXS-101-CL-304 phase 3 study in patients with pre-symptomatic SMA.
Study CL-304 is an ongoing, global, Phase 3, open-label, single-arm, single-dose, multicentre study of intravenous Zolgensma in pre-symptomatic newborn patients up to 6 weeks of age with genetically confirmed bi-allelic SMN1 gene deletions, 2 or 3 copies of the SMN2 gene and absence of the c.859G > C modification in exon 7 of the SMN2 gene. Study CL-304 treated 14 patients with 2 copies of SMN2 and 15 patients with 3 copies of SMN2. At the time of the last study visit prior to 31 December 2019, treated patients with 2 copies of SMN2 were between 6 months and 18.6 months of age and had been in the study for an average of 10.5 months (range: 5.1 to 18.0 months), and the 15 patients with 3 copies of SMN2 were between 3.3 and 15.1 months of age and had been in the study for an average of 8.74 months (range: 2 to 13.9 months). All patients in Study CL-304 were alive and free of permanent ventilation as of their last study visit prior to 31 December 2019. At the time of their last visit before the data cut-off, 6 of the 14 (42.9%) patients with 2 copies of SMN2 were less than 9.2 months of age. The 8 older individuals achieved sitting prior to their last visit before 31 December 2019, at ages ranging from 6.4 to 11.8 months, with 7 of these 8 (87.5%) achieving independent sitting prior to the 9.2 months of age, the 99th percentile for development of this milestone. Twelve patients (85.7%) have achieved CHOP-INTEND scores ≥ 60 as of the 31 December 2019 data cut-off. Of the patients with 3 copies of SMN2, 10 of 15 patients were able to sit without support for at least 30 seconds, 4 patients were able to stand alone without support for at least 3 seconds, and 2 patients were able to walk at least five steps independently. Patients with 3 copies of SMN2 who have not yet achieved the Cohort 2 primary endpoint developmental milestone of standing alone without support for at least 3 seconds are 6.4 to 13.6 months of age. These patients remain within the normal age development window for obtaining these milestones.
5.2 Pharmacokinetic Properties
Pharmacokinetics (PK).
Vector shedding after infusion with onasemnogene abeparvovec was investigated at multiple time points during the completed clinical trial CL-101. Samples of saliva, urine and stool were collected the day after infusion, weekly through Day 30, and then monthly through Month 12 and every 3 months thereafter. Samples from 5 patients were used for onasemnogene abeparvovec vector DNA shedding analysis through the Month 18 visit.
Vector DNA was shed in saliva, urine and stool after infusion of onasemnogene abeparvovec. Clearance was primarily via faeces and the majority was cleared within 30 days after dose administration. The vector DNA concentration in saliva was low on Day 1 after infusion and declined to undetectable levels within 3 weeks. In urine, the vector DNA concentration was very low on Day 1 after infusion and declined to undetectable levels within 1 to 2 weeks. In stool, the vector DNA concentration was much higher than in saliva or urine for 1 to 2 weeks after infusion and declined to undetectable levels by 1 to 2 months after infusion.
Biodistribution.
Biodistribution was evaluated in two patients who died 5.7 months and 1.7 months, respectively, after infusion of onasemnogene abeparvovec at the dose of 1.1 x 1014 vg/kg. Both cases showed that the highest levels of vector DNA were found in the liver. Vector DNA was also detected in the spleen, heart, pancreas, inguinal lymph node, skeletal muscles, peripheral nerves, kidney, lung, intestines, gonads, spinal cord, brain, and thymus. Immunostaining for SMN protein showed generalised SMN expression in spinal motor neurons, neuronal and glial cells of the brain, and in the heart, liver, skeletal muscles, and other tissues evaluated.
5.3 Preclinical Safety Data
In toxicology studies conducted in neonatal mice, dose-dependent cardiac and hepatic toxicities were observed following intravenous administration of onasemnogene abeparvovec. Onasemnogene abeparvovec-related findings in the myocardium, at doses of 7.9 x 1013 vg/kg and higher, included slight to mild mononuclear cell inflammation accompanied by oedema, slight to mild fibrosis, and scattered myocardial cell degeneration/regeneration. Additional cardiac findings at dose levels of 1.5 x 1014 vg/kg and higher included minimal to moderate atrial thrombosis and slight to marked atrial dilation. Liver findings included hepatocellular hypertrophy, Kupffer cell activation, perinuclear vacuolation, and scattered hepatocellular necrosis. Target organ toxicity in the heart and liver was associated with mortality at dose levels of 2.4 x 1014 vg/kg and above, approximately 2.2-fold higher than the recommended clinical dose level.
In a toxicology study conducted in non-human primates, administration of a single dose of onasemnogene abeparvovec intrathecally, without corticosteroid treatment, resulted in minimal to marked mononuclear cell inflammation in the dorsal root ganglia, with neuronal satellitosis, neuronal necrosis, or complete neuronal loss with rare mineralisation. The clinical relevance of this finding is unknown.
Genotoxicity and carcinogenicity.
No studies have been performed to evaluate the genotoxic or carcinogenic potential of onasemnogene abeparvovec.6 Pharmaceutical Particulars
6.1 List of Excipients
Trometamol, magnesium chloride hexahydrate, sodium chloride, poloxamer, hydrochloric acid (for pH adjustment) and water for injections.
Contains no preservative.
6.2 Incompatibilities
In the absence of compatibility studies, this product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
Preparation of pack contents is carried out aseptically under sterile conditions. To reduce microbiological hazard, use as soon as practicable after dispensing into the syringe. If storage is necessary, hold at room temperature (20°C - 25°C) for not more than 6 hours.
6.4 Special Precautions for Storage
Store and transport frozen (≤ -60°C).
Upon receipt, immediately place the pack (in original carton to protect from light) in a refrigerator at 2°C to 8°C.
Zolgensma is stable for 14 days from receipt when stored at 2°C to 8°C.
For storage conditions after thawing of the medicinal product, see Section 6.3 Shelf Life.
Do not refreeze.
6.5 Nature and Contents of Container
Zolgensma is supplied in a single-use vial (10 mL polymer crystal zenith) with stopper (20 mm chlorobutyl rubber) and seal (aluminium, flip-off) with a coloured cap (plastic), in two different vial fill volume sizes, either 5.5 mL or 8.3 mL.
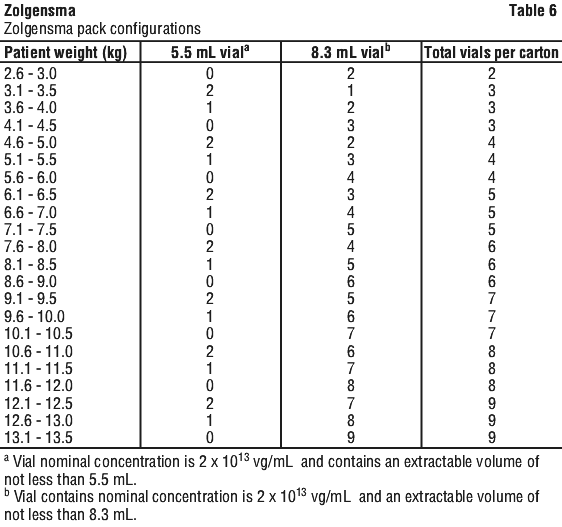
The dose of Zolgensma and exact number of vials required for each patient is calculated according to the patient's weight (see Section 4.2 Dose and Method of Administration; see Table 6).
6.6 Special Precautions for Disposal
This medicine contains genetically modified organisms. Unused medicine and waste products must be disposed of in compliance with the local institutional guidelines for genetically modified organisms or biohazardous waste, as appropriate.
Temporary onasemnogene abeparvovec shedding may occur, primarily through bodily waste. Caregivers and patient families should be advised on the proper handling of patient stools. Instructions should be provided regarding good hand-hygiene when coming into direct contact with patient bodily waste for a minimum of 1 month after onasemnogene abeparvovec treatment. Disposable nappies should be sealed in plastics bags and can be disposed of in household waste.
6.7 Physicochemical Properties
CAS number.
1922968-73-7.7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine.
Summary Table of Changes
