
SUMMARY CMI
CELSENTRI
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I taking CELSENTRI?
CELSENTRI contains the active ingredient maraviroc. CELSENTRI is used in combination with other medicines to treat HIV.
For more information, see Section 1. Why am I using CELSENTRI? in the full CMI.
2. What should I know before I take CELSENTRI?
Do not use if you have ever had an allergic reaction to maraviroc or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I take CELSENTRI? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with CELSENTRI and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take CELSENTRI?
- The usual dosage is either 150 mg, 300 mg or 600 mg twice a day.
- The dose you take will depend on whether you are taking any other medicines with CELSENTRI.
- Swallow the tablets whole with a glass of water. Do not chew.
- CELSENTRI can be taken with or without food
More instructions can be found in Section 4. How do I take CELSENTRI? in the full CMI.
5. What should I know while taking CELSENTRI?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while taking CELSENTRI? in the full CMI.
6. Are there any side effects?
Side effects that have been reported include diarrhoea, constipation, nausea or vomiting, stomach pain or discomfort, indigestion, dizziness, abnormal sense of taste, problems sleeping or abnormal sleep, rash, loss of appetite, muscle spasms or pain, cough, joint pain, fever, colds, upper respiratory tract infections or flu-like symptoms.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
CELSENTRI
Active ingredient(s): maraviroc
Consumer Medicine Information (CMI)
This leaflet provides important information about using CELSENTRI. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using CELSENTRI.
Where to find information in this leaflet:
1. Why am I taking CELSENTRI?
2. What should I know before I take CELSENTRI?
3. What if I am taking other medicines?
4. How do I take CELSENTRI?
5. What should I know while taking CELSENTRI?
6. Are there any side effects?
7. Product details
1. Why am I taking CELSENTRI?
CELSENTRI contains the active ingredient maraviroc. CELSENTRI belongs to a group of medicines called CCR5 blockers.
CELSENTRI is used in combination with other medicines to treat human immunodeficiency virus (HIV).
CELSENTRI reduces the amount of HIV in your body and helps your immune system. It stops the HIV-1 virus entering the CD-4 cells in your blood (also called T-cells). These are the cells in your immune system that the HIV virus attacks.
CELSENTRI works by blocking the most common entry point into the CD-4 cells – called the 'CCR5 receptor'. Because the virus cannot enter the cell, it cannot attack it, and this prevents further damage to your immune system.
CELSENTRI only stops the HIV-1 virus entering the cell, not HIV-2 (another rarer kind of the HIV virus). CELSENTRI also only stops types of HIV-1 virus that enter using the CCR5 receptor. As a result, your doctor would have done a blood test to check what strain of HIV-1 virus you have before they prescribed you this medicine.
You can still pass on HIV when taking this medicine through sexual activity or through passing on blood or bodily secretions which carry the HIV virus.
CELSENTRI does not cure HIV. You should use proper precautions to prevent transmission of HIV to others from occurring. Discuss with your doctor the precautions needed to avoid infecting other people.
2. What should I know before I take CELSENTRI?
Warnings
Do not use CELSENTRI if:
- you are allergic to maraviroc, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any other medical conditions
- have liver problems, hepatitis B or hepatitis C infection so that your liver function can be monitored
- have a history of low blood pressure, low blood pressure upon standing up (known as postural hypotension) or are taking any medicine to lower your blood pressure
- have kidney problems
- have heart problems
- take any medicines for any other condition
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
It is not known if CELSENTRI can harm your unborn child. Your doctor can discuss with you the benefits and risks of taking CELSENTRI whilst pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed. You should not breastfeed if you are infected with HIV because the virus can be transmitted through breastmilk. It is not known whether the active ingredient in CELSENTRI can pass into your breastmilk. Therefore, you should not breastfeed whilst taking CELSENTRI.
Children
- CELSENTRI has not been studied in children less than 18 years of age.
Clinical Trials
More treatment-naïve patients in clinical trials using CELSENTRI had treatment failures and developed resistance to lamivudine compared to patients using efavirenz.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with CELSENTRI and affect how it works.
- efavirenz, etravirine, raltegravir, lopinavir, darunavir, delavirdine, elvitegravir, atazanavir, nelfinavir, indinavir, saquinavir, boceprevir, telaprevir – medicines used to treat HIV or hepatitis C infections
- ketoconazole, itraconazole – medicines used to treat fungal infections
- clarithromycin, telithromycin, rifampicin, rifabutin – medicines used to treat bacterial infections
- medicines containing St John's Wort (hypericum perforatum). St John's Wort can prevent CELSENTRI from working properly. Therefore, you should not take St John's Wort together with CELSENTRI
- carbamazepine, phenobarbital, phenytoin – medicines used to treat seizures
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect CELSENTRI.
4. How do I take CELSENTRI?
How much to take
- The usual dosage is either 150 mg, 300 mg or 600 mg twice a day.
- The dose you take will depend on whether you are taking any other medicines with CELSENTRI. Your doctor will tell you the dose to take and when to take these others medicines.
- Follow the instructions provided and use CELSENTRI until your doctor tells you to stop.
It is important to take all your anti-HIV medicines as prescribed and at the right time of day. This can help your medicines to work better. It also lowers the chance of your medicines becoming less effective in fighting HIV (also known as drug resistance).
How to take CELSENTRI
- Swallow the tablet whole with a drink of water. Do not chew the tablet.
- You can take CELSENTRI with or without food.
If you forget to take CELSENTRI
CELSENTRI should be taken regularly at the same time each day.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise take it as soon as you remember and then go back to taking CELSENTRI as you would normally.
Do not take a double dose to make up for the dose you missed.
If you take too much CELSENTRI
If you think that you have used too much CELSENTRI, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
Symptoms of an overdose may include dizziness or light-headedness when you stand up. If this happens, lie down until you feel better and when you get up, get up slowly.
5. What should I know while taking CELSENTRI?
Things you should do
Call your doctor straight away if you:
- become pregnant whilst taking CELSENTRI
Remind any doctor, dentist or pharmacist you visit that you are using CELSENTRI.
Stay in regular contact with your doctor
CELSENTRI helps to control your condition, but it is not a cure for HIV infection. You need to keep taking it everyday to stop your illness from getting worse. Because CELSENTRI does not cure HIV infections, you may still develop other infections and illnesses linked to HIV.
Things you should not do
- Do not stop using this medicine or change the dose
- Do not start taking any other medicines without first speaking to your doctor
- Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
- Do not use this medicine to treat any other complaints unless your doctor tells you to.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how CELSENTRI affects you.
CELSENTRI may cause dizziness and light-headedness in some people.
Looking after your medicine
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place (below 30°C) away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
Within the first few weeks of treatment with anti-HIV medicines, some people, particularly those that have been HIV positive for some time, may develop inflammatory reactions (e.g. pain, redness, swelling, high temperature) which may resemble an infection and may be severe. It is thought that these reactions are caused by a recovery in the body's ability to fight infections, previously suppressed by HIV.
If you become concerned about any new symptoms, or any changes in your health after starting HIV treatment, discuss with your doctor immediately.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away. |
Very serious side effects
| Very serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Possible chance of infection or cancer
Although there is no evidence from clinical trials of an increase in serious infections or cancer, CELSENTRI affects other immune system cells and therefore may potentially increase your chance of getting other infections or cancer.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What CELSENTRI contains
| Active ingredient (main ingredient) | maraviroc |
| Other ingredients (inactive ingredients) | calcium hydrogen phosphate magnesium stearate microcrystalline cellulose sodium starch glycollate The film-coating (Opadry II Blue (85G20583)) contains: indigo carmine CI73015 macrogol 3350 polyvinyl alcohol soya lecithin talc titanium dioxide |
Do not take this medicine if you are allergic to any of these ingredients.
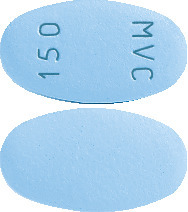
What CELSENTRI looks like
150 mg film-coated tablets are blue in colour, oval shaped and marked "MVC 150" on one side.
Available in blister packs of 60 tablets. AUST R 137329.
300 mg film-coated tablets are blue in colour, oval shaped and marked "MVC 300" on one side.
Available in blister packs of 60 tablets. AUST R 137331
Who distributes CELSENTRI
ViiV Healthcare Pty Ltd
Level 4, 436 Johnston Street
Abbotsford, VIC 3067
Australia
Trademarks are owned by or licenced to the ViiV Healthcare group of companies.
© 2022 ViiV Healthcare group of companies or its licensor.
This leaflet was prepared in October 2022.
Version 9.0.
Published by MIMS December 2022





 MOTIVATE 1 and MOTIVATE 2 were unblinded after the Week 48 visit of the last enrolled patient, and eligible patients could switch to an open-label phase of Celsentri twice daily extending to week 96. A subsequent observational phase extending to 5 years on treatment was conducted to assess the incidence of the following long-term safety selected endpoints; death, AIDS defining events, hepatic failure, MI/cardiac ischemia, malignancies, rhabdomyolysis, and other serious infectious events. The incidence of these selected endpoints was consistent with those seen at earlier time points in the studies and listed in Table 6.
MOTIVATE 1 and MOTIVATE 2 were unblinded after the Week 48 visit of the last enrolled patient, and eligible patients could switch to an open-label phase of Celsentri twice daily extending to week 96. A subsequent observational phase extending to 5 years on treatment was conducted to assess the incidence of the following long-term safety selected endpoints; death, AIDS defining events, hepatic failure, MI/cardiac ischemia, malignancies, rhabdomyolysis, and other serious infectious events. The incidence of these selected endpoints was consistent with those seen at earlier time points in the studies and listed in Table 6.


 Limited numbers of patients from races other than Caucasian were included in the pivotal clinical studies, therefore very limited data are available in these patient populations.
Limited numbers of patients from races other than Caucasian were included in the pivotal clinical studies, therefore very limited data are available in these patient populations.

 The treatment outcomes through week 48 for the treatment-naïve study (MERIT) are shown in Table 13. Treatment outcomes (responders) include reanalysis of the screening samples using a more sensitive tropism assay, enhanced sensitivity Trofile HIV tropism assay, which became available after the Week 48 analysis.
The treatment outcomes through week 48 for the treatment-naïve study (MERIT) are shown in Table 13. Treatment outcomes (responders) include reanalysis of the screening samples using a more sensitive tropism assay, enhanced sensitivity Trofile HIV tropism assay, which became available after the Week 48 analysis. The primary efficacy endpoints were defined as the percentage of patients with HIV-1 RNA undetectable by the standard method (< 400 copies/mL and < 50 copies/mL). After 48 weeks of combination therapy with zidovudine/lamivudine, maraviroc 300 mg BID demonstrated non-inferiority to efavirenz 600 mg QD in the proportion of patients with undetectable viral load measured at < 400 copies/mL but not at < 50 copies/mL (lower bound of 97.5% CI > -10% for non-inferiority). However, reanalysis of the data following rescreening of the samples using the enhanced sensitivity tropism assay demonstrated non-inferiority for maraviroc 300 mg BID compared to efavirenz 600 mg QD in the proportion of patients with viral loads of < 400 copies/mL and < 50 copies/mL.
The primary efficacy endpoints were defined as the percentage of patients with HIV-1 RNA undetectable by the standard method (< 400 copies/mL and < 50 copies/mL). After 48 weeks of combination therapy with zidovudine/lamivudine, maraviroc 300 mg BID demonstrated non-inferiority to efavirenz 600 mg QD in the proportion of patients with undetectable viral load measured at < 400 copies/mL but not at < 50 copies/mL (lower bound of 97.5% CI > -10% for non-inferiority). However, reanalysis of the data following rescreening of the samples using the enhanced sensitivity tropism assay demonstrated non-inferiority for maraviroc 300 mg BID compared to efavirenz 600 mg QD in the proportion of patients with viral loads of < 400 copies/mL and < 50 copies/mL. The median increase from baseline in CD4+ cell counts at week 96 was 184 cells/mm3 for the Celsentri arm compared to 155 cells/mm3 for the efavirenz arm.
The median increase from baseline in CD4+ cell counts at week 96 was 184 cells/mm3 for the Celsentri arm compared to 155 cells/mm3 for the efavirenz arm.
