1 Name of Medicine
Alendronate sodium trihydrate.
2 Qualitative and Quantitative Composition
Each tablet contains alendronate sodium trihydrate equivalent to 70 mg alendronic acid.
3 Pharmaceutical Form
Tablets.
Alendronate-WGR Once Weekly 70 mg tablets.
White, oval, biconvex tablets, engraved APO on one side and ALE 70 on the reverse.4.1 Therapeutic Indications
Treatment of osteoporosis. Osteoporosis must be confirmed by the finding of low bone mass of at least two standard deviations below the gender specific mean for young adults, or by the presence of osteoporotic fracture.
4.2 Dose and Method of Administration
Alendronate-WGR Once Weekly 70 mg tablets are intended for oral administration.
Dosage.
Treatment of osteoporosis.
The recommended dosage is one Alendronate Once Weekly (70 mg) tablet once weekly.
Alendronate must be taken at least 30 minutes before the first food, beverage or medication of the day with plain water only. Other beverages (including mineral water), food and some medications are likely to reduce the absorption of alendronate (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Alendronate should only be taken upon arising for the day. To facilitate delivery to the stomach and thus reduce the potential for oesophageal irritation, an alendronate tablet should be swallowed with a full glass of water. Patients should not crush, chew or suck on the tablet because of a potential for stomatitis or oropharyngeal ulceration.
Patients should not lie down for at least 30 minutes and until after their first food of the day. Alendronate should not be taken at bedtime or before arising for the day. Failure to follow these instructions may increase the risk of oesophageal adverse experiences (see Section 4.4 Special Warnings and Precautions for Use).
Severe oesophageal ulceration has been reported in patients taking this drug (see Section 4.4 Special Warnings and Precautions for Use). Patients should be instructed that if they develop symptoms of oesophageal disease (such as difficulty or pain upon swallowing, retrosternal pain or new or worsening heartburn) they should stop taking alendronate and consult their physician.
In clinical trials, alendronate was administered with appropriate calcium, and vitamin D supplementation. The use of vitamin D as the sole treatment of osteoporosis has not been established.
Patients should receive supplemental calcium and vitamin D if dietary intake is inadequate (see Section 4.4 Special Warnings and Precautions for Use). Physicians should consider the vitamin D intake from vitamins and dietary supplements. Additional supplements should not be taken at the same time of day as alendronate (see above).
Although no specific studies have been conducted on the effects of switching patients on another therapy for osteoporosis to alendronate, there are no known or theoretical safety concerns related to alendronate in patients who previously received any other antiosteoporotic therapy.
Renal impairment.
No dosage adjustment is necessary for the elderly or patients with mild to moderate renal insufficiency (creatinine clearance 35 to 60 mL/minute). Alendronate is not recommended for patients with more severe renal insufficiency (creatinine clearance < 35 mL/minute).4.3 Contraindications
Abnormalities of the oesophagus which delay oesophageal emptying, such as stricture or achalasia.
Inability to stand or sit upright for at least 30 minutes.
Hypersensitivity to any component of this product.
Hypocalcaemia (see Section 4.4 Special Warnings and Precautions for Use).
4.4 Special Warnings and Precautions for Use
Severe oesophageal ulceration has been reported in patients taking alendronate (see Section 4.2 Dose and Method of Administration). Physicians should therefore be alert to any signs or symptoms signalling a possible oesophageal reaction. Patients should be instructed to discontinue alendronate and seek medical attention if they develop dysphagia, odynophagia or retrosternal pain.
General.
Causes of osteoporosis other than hypogonadism, ageing and glucocorticoid use should be considered.
If there are clinical reasons to suspect hypocalcaemia and/or vitamin D deficiency (serum levels 25-hydroxyvitamin D < 9 nanomol/L), the appropriate diagnostic tests should be performed. Hypocalcaemia must be corrected before initiating therapy with alendronate (see Section 4.3 Contraindications). Other disturbances of mineral metabolism, e.g. vitamin D deficiency, should also be effectively treated. In patients with these conditions, serum calcium and symptoms of hypocalcaemia should be monitored during therapy with alendronate.
Small, asymptomatic decreases in serum calcium and phosphate may occur, especially in patients with Paget's disease, in whom the pretreatment rate of bone turnover may be greatly elevated, and in patients receiving glucocorticoids, in whom calcium absorption may be decreased.
Ensuring adequate calcium and vitamin D intake is especially important in patients with Paget's disease of bone and in patients receiving glucocorticoids.
In double blind, multicentre controlled studies, asymptomatic, mild and transient decreases in serum calcium and phosphate were observed in approximately 18 and 10%, respectively, of patients taking alendronate versus approximately 12 and 3% of those taking placebo. However, the incidences of decreases in serum calcium to < 8.0 mg/dL (2.0 mM) and serum phosphate to ≤ 2.0 mg P/dL (0.65 mM) were similar in both treatment groups.
Alendronate, like other bisphosphonates, may cause local irritation of the upper gastrointestinal mucosa.
Oesophageal adverse experiences such as oesophagitis, oesophageal ulcers and oesophageal erosions, rarely followed by oesophageal stricture or perforation, have been reported in patients receiving treatment with alendronate. In some cases, these have been severe and required hospitalisation.
The risk of severe oesophageal adverse experiences appears to be greater in patients who lie down after taking alendronate and/or who fail to swallow it with a full glass of water, and/or who continue to take alendronate after developing symptoms suggestive of oesophageal irritation. Therefore, it is very important that the full dosing instructions are provided to, and understood by, the patient (see Section 4.2 Dose and Method of Administration).
While no increased risk was observed in extensive clinical trials, there have been rare (post-marketing) reports of gastric and duodenal ulcers, some severe and with complications.
Because of possible irritant effects of alendronate on the upper gastrointestinal mucosa and a potential for worsening of the underlying disease, caution should be used when alendronate is given to patients with active upper gastrointestinal problems such as dysphagia, oesophageal diseases (including known Barrett's Oesophagus), gastritis, duodenitis or ulcers.
Dental.
Localised osteonecrosis of the jaw (ONJ), generally associated with tooth extraction and/or local infection (including osteomyelitis) with delayed healing, has been reported rarely with oral bisphosphonates including alendronate (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience).
As of May 2004, ONJ after bisphosphonate treatment has been described in a total of 99 cases in two large case series, 7 of which were taking oral bisphosphonates. As of 3 Nov 2006, the Australian Adverse Drug Reactions Advisory Committee has received 25 reports of ONJ in patients receiving alendronate.
Most reported cases of bisphosphonate-associated ONJ have been in cancer patients treated with intravenous bisphosphonates. Known risk factors for ONJ include a diagnosis of cancer, concomitant therapies (e.g. chemotherapy, radiotherapy, corticosteroids, angiogenesis inhibitors), poor oral hygiene, co-morbid disorders (e.g. periodontal and/or other pre-existing dental disease, anaemia, coagulopathy, infection) and smoking.
A dental examination with appropriate preventative dentistry should be considered prior to treatment with bisphosphonates in patients with possible risk factors.
Before commencing invasive dental procedures, patients and their dentists should be advised of the risk of osteonecrosis of the jaw so that any symptoms, including toothache, developing during treatment can be fully assessed for cause before treatment of the tooth commences.
For patients requiring invasive dental surgery (e.g. tooth extraction, dental implants), there are no data available to suggest whether discontinuation of bisphosphonate treatment reduces the risk of ONJ. Therefore, clinical judgement of the treating physician and/or oral surgeon should guide the management plan, including discontinuation of bisphosphonate treatment, of each patient based on individual benefit/risk assessment.
In patients who develop ONJ while on bisphosphonate therapy, the clinical judgement of the treating physician should guide the management plan to include appropriate care by an oral surgeon and discontinuation of bisphosphonate therapy should be based on individual benefit/risk assessment.
Surgery at the affected area may exacerbate the condition.
Atypical stress fractures.
A small number of long-term (usually longer than three years) alendronate-treated patients developed stress fractures of the subtrochanteric and proximal femoral shaft and other bones. Some were stress fractures (some of which were reported as insufficiency fractures) occurring in the absence of apparent trauma or induced by mild external force. Fractures are often bilateral; therefore, the contralateral femur should be examined in patients who have sustained a femoral shaft stress fracture. Poor healing of these fractures was also reported.
Some patients experienced prodromal pain in the affected area, often associated with imaging features of stress fracture, weeks to months before a complete fracture occurred. The number of reported cases of this condition is very low (some 40 reported cases worldwide). Approximately one third of the reported femur fractures were bilateral; therefore the contralateral femur should be examined in patients who have sustained a femoral shaft stress fracture. Patients with suspected stress fractures should be evaluated, including evaluation for known causes and risk factors (e.g. vitamin D deficiency, malabsorption, glucocorticoid use, previous stress fracture, lower extremity arthritis or fracture, extreme or increased exercise, diabetes mellitus, chronic alcohol abuse), and receive appropriate orthopaedic care. Discontinuation of bisphosphonate therapy in patients with stress fractures is advisable pending evaluation of the patient, based on individual benefit/risk assessment. A cause and effect relationship between bisphosphonate use and stress fractures has not been excluded.
Musculoskeletal pain.
Bone, joint, and/or muscle pain has been reported in patients taking bisphosphonates. In post-marketing experience, these symptoms have rarely been severe and/or incapacitating (see Section 4.8 Adverse Effects (Undesirable Effects), Post-marketing experience). The time to onset of symptoms varied from one day to several months after starting treatment. Most patients had relief of symptoms after stopping treatment. A subset had recurrence of symptoms when rechallenged with the same drug or another bisphosphonate.
Adynamic bone disease.
Severely depressed bone turnover has been reported in connection with long-term use, manifesting as delayed or absent fracture healing. All patients had spontaneous atraumatic nonspinal fractures. These patients had received alendronate for three to nine years at doses of 10 mg/day or 70 mg/week. Three women had received estrogen therapy. Histomorphometric analysis of the cancellous bone in nine patients (eight women, one man) showed suppressed bone formation with reduced or absent osteoblastic surface in most patients, low or normal osteoclastic surface in eight patients and eroded surface was decreased in four patients. Four of nine patients showed satisfactory fracture healing within eight months. No new fractures occurred after cessation of alendronate. The presence of spontaneous non-spinal fractures, e.g. of femoral shaft, in patients who have received alendronate long-term may be indicative of adynamic bone disease.
Nephrolithiasis and hypercalciuria.
Patients with a history of either nephrolithiasis or hypercalciuria may require special diets that limit their calcium intake.
Dosing instructions for patients.
To facilitate delivery to the stomach and thus reduce the potential for oesophageal irritation, patients should be instructed to swallow each tablet of alendronate with a full glass of water. Patients should be instructed not to lie down for at least 30 minutes and until after their first food of the day. Patients should not crush, chew or suck on the tablet because of a potential for stomatitis or oropharyngeal ulceration.
Patients should be specifically instructed not to take alendronate at bedtime or before arising for the day. Patients should be informed that failure to follow these instructions may increase their risk of oesophageal problems. Patients should be instructed that if they develop symptoms of oesophageal disease, e.g. difficulty or pain upon swallowing, retrosternal pain or new or worsening heartburn, they should stop taking alendronate and consult their doctor.
Patients should be instructed that if they miss a dose of alendronate once weekly (70 mg), they should take one tablet on the morning after they remember. They should not take two tablets on the same day but should return to taking one tablet once a week, as originally scheduled on their chosen day.
Use in renal impairment.
Alendronate is not recommended for patients with creatinine clearance less than 35 mL/minute (see Section 4.2 Dose and Method of Administration).
Use in the elderly.
In controlled trials, there was no age related difference in the efficacy or safety profiles of alendronate.
Paediatric use.
Alendronate has not been studied in children and should not be given to them.
Effects on laboratory tests.
In double-blind, multicentre controlled studies, asymptomatic, mild and transient decreases in serum calcium and phosphate were observed in approximately 18 and 10%, respectively, of patients taking alendronate versus approximately 12 and 3% of those taking placebo. However, the incidences of decreases in serum calcium to < 8.0 mg/dL (2.0 mM) and serum phosphate to ≤ 2.0 mg P/dL (0.65 mM) were similar in both treatment groups.4.5 Interactions with Other Medicines and Other Forms of Interactions
If taken at the same time, it is likely that calcium supplements, antacids and other oral medications will interfere with the absorption of alendronate. Therefore, patients must wait at least one-half hour after taking alendronate before taking any other oral medication.
No other drug interactions of clinical significance are anticipated although the concomitant medication with two or more bisphosphonates cannot be recommended because of the lack of clinical data.
Concomitant use of hormone replacement therapy (HRT) (estrogen ± progestin) and alendronate was assessed in two clinical studies of one or two years duration in post-menopausal osteoporotic women. Combined use of alendronate and HRT resulted in greater increases in bone mass, together with greater decreases in bone turnover, than seen with either treatment alone. In these studies, the safety and tolerability profile of the combination was consistent with those of the individual treatments (see Section 4.8 Adverse Effects (Undesirable Effects), Clinical studies, Concomitant use with estrogen/ hormone replacement therapy).
Specific interaction studies were not performed. Alendronate (10 and 5 mg/day) was used in studies of treatment and prevention of osteoporosis in post-menopausal women, men and glucocorticoid users, with a wide range of commonly prescribed drugs without evidence of clinical adverse interactions. In clinical studies, the incidence of upper gastrointestinal adverse events was increased in patients receiving daily therapy with dosages of alendronate greater than 10 mg and aspirin containing products. However, this was not observed in studies with alendronate once weekly (70 mg).
Since nonsteroidal anti-inflammatory drug (NSAID) use is associated with gastrointestinal irritation, caution should be used during concomitant use with alendronate.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Respective oral alendronate doses of up to 9 and 15 mg/kg/day had no effect on fertility in male and female rats.
(Category B3)
Alendronate has not been studied in pregnant women and should not be given to them. In studies with pregnant rats, oral doses of alendronate 2 mg/kg/day and above resulted in dystocia due to maternal hypocalcaemia. Foetal weight was reduced in rats at maternal doses greater than 5 mg/kg/day. No teratogenic effects were seen in rats or rabbits at oral doses up to 25 and 35 mg/kg/day, respectively.
Alendronate has not been studied in breastfeeding women and should not be given to them.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects on the ability to drive and use machines have been performed. However, certain adverse reactions that have been reported with alendronate may affect some patients' ability to drive or operate machinery. Individual responses to alendronate may vary (see Section 4.8 Adverse Effects (Undesirable Effects)).
4.8 Adverse Effects (Undesirable Effects)
Clinical studies.
In clinical studies alendronate was generally well tolerated. In studies up to five years in duration, side effects, which usually were mild, generally did not require discontinuation of therapy.
Treatment of osteoporosis.
Post-menopausal women.
Alendronate has been evaluated for safety in clinical studies in approximately 5000 post-menopausal patients. In two three-year placebo controlled double blind multicentre studies, discontinuation of therapy due to any clinical adverse experience occurred in 4.1% of 196 patients treated with alendronate 10 mg/day and 6.0% of 397 patients treated with placebo. Adverse experiences reported by the investigators as possibly, probably or definitely drug related in ≥ 1% of patients treated with either alendronate 10 mg/day or placebo are presented in Table 1.
 Rarely, rash and erythema have occurred.
Rarely, rash and erythema have occurred.
In the two year extension (treatment years 4 and 5) of the above studies, the overall safety profile of alendronate 10 mg/day was similar to that observed during the three year placebo controlled period. Additionally, the proportion of patients who discontinued alendronate 10 mg/day due to any clinical adverse experience was similar to that during the first three years of the study.
In the Fracture Intervention Trial, discontinuation of therapy due to any clinical adverse experience occurred in 9.1% of 3236 patients treated with alendronate 5 mg/day for two years and 10 mg/day for either one or two additional years and 10.1% of 3223 patients treated with placebo. Discontinuations due to upper gastrointestinal adverse experiences were: alendronate 3.2%; placebo 2.7%. The overall adverse experience profile was similar to that seen in other studies with alendronate 5 or 10 mg/day.
In a one year, double blind, multicentre study, the overall safety and tolerability profiles of alendronate once weekly (70 mg) (n=519) and alendronate 10 mg daily (n=370) were similar. Adverse experiences reported by the investigators as possibly, probably or definitely drug related in greater than or equal to 1% of patients treated with either patient group are presented in Table 2.

Concomitant use with estrogen/ hormone replacement therapy.
In two studies (of one and two years duration) of post-menopausal osteoporotic women (total n = 853), the safety and tolerability profile of combined treatment with alendronate 10 mg once daily and estrogen ± progestin (n=354) was consistent with those of the individual treatments.
Men.
In a two year, placebo controlled, double blind, multicentre study, the safety profile of alendronate 10 mg daily in 146 men was generally similar to that seen in post-menopausal women.
Other studies in men and women.
In a ten week endoscopy study in men and women (n=277; mean age 55 years) no difference was seen in upper gastrointestinal tract lesions between alendronate once weekly 70 mg and placebo.
In an additional one year study in men and women (n=335; mean age 50 years) the overall safety and tolerability profiles of alendronate once weekly 70 mg were similar to that of placebo and no difference was seen between men and women.
Treatment and prevention of glucocorticoid-induced osteoporosis.
In two, one-year, placebo-controlled, double-blind, multicentre studies in patients receiving glucocorticoid treatment, the overall safety and tolerability profiles of alendronate 5 and 10 mg/day were generally similar to that of the placebo. Adverse experiences reported by the investigators as possible, probably or definitely drug related in equal or more than 1% of patients treated with either alendronate 5 mg/day, 10 mg/day or placebo are presented in Table 3.

Post-marketing experience.
The following adverse reactions have been reported in post-marketing use.
Body as a whole.
Hypersensitivity reactions including urticaria and, rarely, angioedema. Transient symptoms as in an acute phase response (myalgia, malaise, asthenia and, rarely, fever) have been reported with alendronate, typically in association with initiation of treatment. Rarely, symptomatic hypocalcaemia has occurred, generally in association with predisposing conditions. Rarely, peripheral oedema.
Gastrointestinal.
Nausea, vomiting, oesophagitis, oesophageal erosions, oesophageal ulcers, rarely oesophageal stricture or perforation, and oropharyngeal ulceration and/or stomatitis; rarely, gastric or duodenal ulcers, some severe and with complications (see Section 4.4 Special Warnings and Precautions for Use; Section 4.2 Dose and Method of Administration).
Musculoskeletal.
Bone, joint, and/or muscle pain, rarely severe and/or incapacitating (see Section 4.4 Special Warnings and Precautions for Use); joint swelling, atypical stress fracture, low-energy fractures of the femoral shaft and other bones (see Section 4.4 Special Warnings and Precautions for Use).
Localised osteonecrosis of the jaw, generally associated with tooth extraction and/or local infection (including osteomyelitis), often with delayed healing, has been reported rarely.
Nervous system.
Dizziness, vertigo, dysgeusia.
Skin.
Rash (occasionally with photosensitivity), pruritus, alopecia, rarely severe skin reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis.
Special senses.
Rarely uveitis, scleritis or episcleritis. Cholesteatoma of the external auditory canal (focal osteonecrosis) has been reported rarely.
Bioequivalence study.
Chest pain, erythema and dry skin, and pyrexia were reported during the bioequivalence study and classified as probably related to the study drug.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Symptoms.
No specific information is available on the treatment of overdose with alendronate. Hypocalcaemia, hypophosphataemia and upper gastrointestinal adverse events such as upset stomach, heartburn, oesophagitis, gastritis or ulcer may result from oral overdosage.
Treatment.
Administration of milk or antacids to bind alendronate should be considered.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Alendronate is a bisphosphonate that, in animal studies, localises preferentially to sites of bone resorption, specifically under osteoclasts, and inhibits osteoclastic bone resorption with no direct effect on bone formation. Since bone formation and bone resorption are coupled, bone formation is also reduced, but less so than resorption, leading to progressive gains in bone mass (see Clinical trials, below). Following exposure to alendronate, normal bone is formed that incorporates alendronate into its matrix, where it is pharmacologically inactive.
The relative inhibitory activities on bone resorption and mineralisation of alendronate and etidronate were compared in growing rats. The lowest dose of alendronate that interfered with bone mineralisation (leading to osteomalacia) was 6000-fold the anti-resorptive dose. The corresponding safety margin for etidronate was one to one. These data indicate that, unlike etidronate, alendronate administered in therapeutic doses is highly unlikely to induce osteomalacia.
Osteoporosis.
The World Health Organization (WHO) utilises the definition of osteoporosis as a disease characterised by low bone mass and microarchitectural deterioration of bone tissue, leading to enhanced bone fragility and a consequent increase in fracture risk. The diagnosis may be confirmed by the finding of low bone mass (e.g. at least two standard deviations below the gender specific mean for young adults) or by the presence or history of osteoporotic fracture. It occurs in both males and females but is most common among women following the menopause, when bone turnover increases and the rate of bone resorption exceeds that of bone formation, leading to loss of bone mass.
Osteoporosis in post-menopausal women.
Daily oral doses of alendronate in post-menopausal women produced biochemical changes indicative of dose dependent inhibition of bone resorption, including decreases in urinary calcium and urinary markers of bone collagen degradation (such as hydroxyproline, deoxypyridinoline and cross linked N-telopeptides of type I collagen). These biochemical changes returned toward baseline values as early as three weeks following the discontinuation of alendronate despite the long retention of alendronate in the skeleton.
Long-term treatment of osteoporosis with alendronate 10 mg/day (for up to five years) reduced urinary excretion of markers of bone resorption, deoxypyridinoline and cross linked N-telopeptides of type I collagen by approximately 50 and 70%, respectively, to reach levels similar to those seen in healthy premenopausal women. Similar decreases were seen in patients in osteoporosis prevention studies who received alendronate 5 mg/day. The decrease in the rate of bone resorption indicated by these markers was evident as early as one month and at three to six months reached a plateau that was maintained for the entire duration of treatment with alendronate. In osteoporosis treatment studies alendronate 10 mg/day decreased the markers of bone formation, osteocalcin and total serum alkaline phosphatase, by approximately 50% and 25 to 30%, respectively, to reach a plateau after 6 to 12 months. Similar though slightly lower reductions in the rate of bone turnover were observed in post-menopausal women during one year studies with alendronate once weekly (70 mg) for the treatment of osteoporosis.
Osteoporosis in men.
Even though osteoporosis is less prevalent in men than in post-menopausal women, a significant proportion of osteoporotic fractures occur in men. The prevalence of vertebral deformities appears to be similar in men and women. All men with osteoporosis should be investigated for hypogonadism and, if necessary, treated for this condition. Treatment of men with osteoporosis with alendronate 10 mg/day for two years reduced urinary excretion of cross linked N-telopeptides of type I collagen by approximately 60% and bone specific alkaline phosphatase by approximately 40%. Similar reductions in cross linked N-telopeptides of type I collagen were seen in men receiving alendronate 70 mg once weekly.
Clinical trials.
Treatment of osteoporosis.
Post-menopausal women. Effect on bone mineral density.
The efficacy of alendronate 10 mg once daily in post-menopausal women with osteoporosis was demonstrated in two large three year multicentre studies of virtually identical design, one performed in the United States and the other in 15 different countries (multinational), which enrolled 478 and 516 patients, respectively. Figure 1 shows the mean increases in bone mineral density (BMD) of the lumbar spine, femoral neck and trochanter in patients receiving alendronate 10 mg/day relative to placebo treated patients at three years for each of these studies.
 These increases were highly significant relative both to baseline and placebo at each measurement site in each study. Increases in BMD were evident as early as three months and continued throughout the entire three years of treatment. See Figure 2 for lumbar spine results. In the two year extension of these studies, treatment with alendronate 10 mg/day resulted in continued increases in BMD at the lumbar spine and trochanter (absolute additional increases between years 3 and 5: lumbar spine 0.94%; trochanter 0.88%). BMD at the femoral neck, forearm and total body were maintained. Thus, alendronate appears to reverse the progression of osteoporosis as assessed by increased bone mineral density. Alendronate was similarly effective regardless of age, race, baseline rate of bone turnover, renal function and use of concomitant medications.
These increases were highly significant relative both to baseline and placebo at each measurement site in each study. Increases in BMD were evident as early as three months and continued throughout the entire three years of treatment. See Figure 2 for lumbar spine results. In the two year extension of these studies, treatment with alendronate 10 mg/day resulted in continued increases in BMD at the lumbar spine and trochanter (absolute additional increases between years 3 and 5: lumbar spine 0.94%; trochanter 0.88%). BMD at the femoral neck, forearm and total body were maintained. Thus, alendronate appears to reverse the progression of osteoporosis as assessed by increased bone mineral density. Alendronate was similarly effective regardless of age, race, baseline rate of bone turnover, renal function and use of concomitant medications.
 In patients with post-menopausal osteoporosis treated with alendronate 10 mg/day for one or two years the effects of treatment withdrawal were assessed. Following discontinuation, there were no further increases in bone mass and the rates of bone loss were similar to those in the placebo groups. These data indicate that continuous treatment with alendronate is required to produce progressive increases in bone mass.
In patients with post-menopausal osteoporosis treated with alendronate 10 mg/day for one or two years the effects of treatment withdrawal were assessed. Following discontinuation, there were no further increases in bone mass and the rates of bone loss were similar to those in the placebo groups. These data indicate that continuous treatment with alendronate is required to produce progressive increases in bone mass.
The therapeutic equivalence of alendronate once weekly (70 mg) (n = 519) and alendronate 10 mg daily (n=370) was demonstrated in a one year, double blind, multicentre study of post-menopausal women with osteoporosis. The mean increases from baseline in lumbar spine BMD at one year were 5.1% (4.8, 5.4%; 95% CI) in the 70 mg once weekly group and 5.4% (5.0, 5.8%; 95% CI) in the 10 mg daily group. The two treatment groups were also similar with regard to BMD increases at other skeletal sites. While there are no placebo controlled fracture data for the alendronate once weekly (70 mg) tablet, the increases in bone density support the expectation that alendronate once weekly (70 mg) will have effects to reduce the incidence of fractures similar to those of the 10 mg daily treatment (see below). The study was not designed to evaluate the relative compliance of alendronate once weekly (70 mg) and 10 mg daily.
Effect on fracture incidence.
Although the US and multinational studies (see above) were not designed to assess fracture rates as the primary endpoint, preplanned analysis of the data pooled across once daily doses at three years revealed a statistically significant and clinically meaningful 48% reduction in the proportion of patients treated with alendronate experiencing one or more vertebral fractures (3.2%) relative to those treated with placebo (6.2%). Furthermore, of patients who sustained any vertebral fracture, those treated with alendronate experienced less height loss (5.9 versus 23.3 mm) due to a reduction in both the number and severity of fractures.
The Fracture Intervention Trial (FIT) consisted of two studies in post-menopausal women: the three year study of patients who had at least one baseline vertebral (compression) fracture and the four year study of patients with low bone mass but without baseline vertebral fracture.
Fracture intervention trial: three year study (patients with at least one baseline vertebral fracture).
This randomised, double blind, placebo controlled 2027 patient study, (alendronate n=1022; placebo, n=1005) demonstrated that treatment with alendronate resulted in clinically significant reductions in fracture incidence at three years as shown in Table 4. Data also showed statistically significant reductions in painful vertebral fractures and clinical fractures at other sites. Similar reductions of hip and wrist fractures were seen in five pooled osteoporosis treatment studies of two or three years duration.
 Furthermore, in this population of patients with baseline vertebral fracture, treatment with alendronate significantly reduced the incidence of hospitalisations resulting from any cause (25.0% versus 30.7%, a 20% relative risk reduction). This difference appears to be related, at least in part, to the reduction in fracture incidence.
Furthermore, in this population of patients with baseline vertebral fracture, treatment with alendronate significantly reduced the incidence of hospitalisations resulting from any cause (25.0% versus 30.7%, a 20% relative risk reduction). This difference appears to be related, at least in part, to the reduction in fracture incidence.
Fracture intervention trial: four year study (patients with low bone mass but without a baseline vertebral fracture).
This randomised, double blind, placebo controlled, 4432 patient study (alendronate, n=2214; placebo, n=2218) further demonstrated the reduction in fracture incidence due to alendronate. The intent of the study was to recruit women with osteoporosis, i.e. with a baseline femoral neck BMD at least two standard deviations below the mean for young adult women. However, due to subsequent revisions to the normative values for femoral neck BMD, 31% of patients were found not to meet this entry criterion and thus this study included both osteoporotic and nonosteoporotic women. The results are shown in Table 5 for the patients with osteoporosis.

Consistency of fracture results.
The reductions in the incidence of vertebral fractures (alendronate versus placebo) in the three and four year studies of FIT were consistent with that in the combined US and multinational (US/Mult) treatment studies (see above), in which 80% of the women did not have a vertebral fracture at baseline. During these studies, treatment with alendronate reduced the proportion of women experiencing at least one new vertebral fracture by approximately 50% (three year FIT: 47% reduction, p < 0.001; four year FIT: 44% reduction, p=0.001 US/Mult, 48% reduction, p=0.034). In addition, alendronate reduced the proportion of women experiencing multiple (two or more) new vertebral fractures by approximately 90% in the US/Mult and three year FIT studies (p < 0.001). Thus, alendronate reduced the incidence of vertebral fractures whether or not patients had experienced a previous vertebral fracture.
Overall, these results demonstrate the consistent efficacy of alendronate in reducing the incidence of fractures, including those of the spine and hip, which are the sites of osteoporotic fracture associated with greatest morbidity.
Bone histology.
Bone histology in 270 post-menopausal patients with osteoporosis treated with alendronate at doses ranging from 1 to 20 mg/day for one, two or three years revealed normal mineralisation and structure, as well as the expected decrease in bone turnover relative to placebo. These data, together with the normal bone histology and increased bone strength observed in ovariectomised rats and baboons exposed to long-term alendronate treatment, indicate that bone formed during therapy with alendronate is of normal quality.
Concomitant use with estrogen/ hormone replacement therapy.
The effects on BMD of treatment with alendronate 10 mg once daily and conjugated estrogen (0.625 mg/day) either alone or in combination were assessed in a two year, double blind, placebo controlled study of hysterectomised post-menopausal osteoporotic women (n=425). At two years, the increases in lumbar spine BMD from baseline were significantly greater with the combination (8.3%) than with either estrogen or alendronate alone (both 6.0%).
The effects on BMD when alendronate was added to stable doses (for at least one year) of HRT (estrogen ± progestin) were assessed in a one year, double blind, placebo controlled study in post-menopausal osteoporotic women (n=428). The addition of alendronate 10 mg once daily to HRT produced, at one year, significantly greater increases in lumbar spine BMD (3.7%) versus HRT alone (1.1%).
In these studies, significant increases or favourable trends in BMD for combined therapy compared with HRT alone were seen at the total hip, femoral neck and trochanter. No significant effect was seen for total body BMD.
Men.
The efficacy of alendronate 10 mg once daily in men with osteoporosis was demonstrated in a two year, double blind, placebo controlled, multicentre study, which enrolled 241 osteoporotic men between the ages of 31 and 87 years. All patients in the study (97.5% of whom were Caucasian) had either: 1) a BMD T-score less than or equal to -2 at the femoral neck and less than or equal to -1 at the lumbar spine or 2) a baseline osteoporotic fracture and a BMD T-score of less than or equal to -1 at the femoral neck. At two years the mean increases relative to placebo in BMD in men receiving alendronate 10 mg daily were: lumbar spine 5.3%; femoral neck 2.6%; trochanter 3.1%; and total body 1.6% (all p less than or equal to 0.001). Alendronate was effective regardless of age, gonadal function, baseline rate of bone turnover, or baseline BMD. Consistent with the much larger studies in post-menopausal women, in these men alendronate 10 mg daily reduced the incidence of new vertebral fracture (post hoc analysis; assessment by quantitative radiography) relative to placebo (0.8 versus 7.1%, respectively; p = 0.017) and, correspondingly, also reduced height loss (-0.6 versus -2.4 mm, respectively; p=0.022).
The effects of discontinuation of alendronate treatment have not been studied in this population.
5.2 Pharmacokinetic Properties
Absorption.
Relative to an intravenous reference dose, the mean oral bioavailability of alendronate in women was 0.64% for doses ranging from 5 to 70 mg when administered after an overnight fast and two hours before a standardised breakfast. There was substantial variability both within and between patients, coefficient of variation 63 and 77%, respectively. Oral bioavailability in men (0.6%) was similar to that in women.
Bioavailability was decreased similarly, (by approximately 40%) whether alendronate was administered one hour or 30 minutes before a standardised breakfast. In osteoporosis and Paget's disease studies, alendronate was effective when administered at least 30 minutes before the first food or beverage of the day.
Bioavailability was negligible whether alendronate was administered with or up to two hours after a standardised breakfast. Concomitant administration of alendronate with coffee or orange juice reduced bioavailability by approximately 60%.
In normal subjects, oral prednisone (20 mg three times daily for five days) did not substantially alter the oral bioavailability of alendronate (alendronate alone: 0.73%; alendronate plus prednisone: 0.87%).
Distribution.
Pre-clinical studies show that alendronate transiently distributes to soft tissues following administration but is then rapidly redistributed to bone or excreted in the urine. The mean steady-state volume of distribution, exclusive of bone, is at least 28 L in humans. Concentrations of alendronate in plasma following therapeutic oral doses are generally below the limits of quantification (< 5 nanogram/mL). Protein binding in human plasma is approximately 78%.
Metabolism.
There is no evidence that alendronate is metabolised in animals or humans.
Excretion.
Following a single intravenous dose of C-alendronate 10 mg, approximately 50% of the radioactivity was excreted in the urine within 72 hours and little or no radioactivity was recovered in the faeces; the renal clearance of alendronate was 71 mL/minute. Plasma concentrations fell by more than 95% within six hours following intravenous administration, due to distribution to the bone and excretion in the urine. The terminal half-life in humans is estimated to exceed ten years, reflecting release of alendronate from the skeleton. Alendronate is not excreted through the acidic or basic transport systems of the kidney in rats, and thus it is not anticipated to interfere with the excretion of other drugs by those systems in humans.
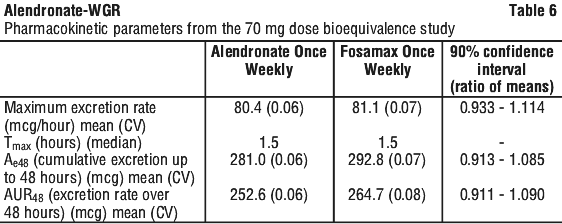
One bioequivalence study, specific to alendronate once weekly, was conducted comparing alendronate once weekly with Fosamax Once Weekly tablets in 127 subjects. When administered as a single oral dose of 70 mg, urinary excretion rates were the same for both formulations (see Table 6). Thus, alendronic acid tablets are shown to be bioequivalent with Fosamax and may be used interchangeably. The acceptance criteria for the confidence intervals of the pharmacokinetic parameters in this study was 0.80 to 1.25.
 Preclinical studies show that any drug that is not deposited in bone is rapidly excreted in the urine. No evidence of saturation of bone uptake was found over three weeks in rats with a cumulative intravenous dose of 35 mg/kg. Although no clinical information is available, it is likely that, as in animals, elimination of alendronate via the kidney will be reduced in patients with impaired renal function. Therefore, somewhat greater accumulation of alendronate in bone might be expected in patients with impaired renal function (see Section 4.2 Dose and Method of Administration).
Preclinical studies show that any drug that is not deposited in bone is rapidly excreted in the urine. No evidence of saturation of bone uptake was found over three weeks in rats with a cumulative intravenous dose of 35 mg/kg. Although no clinical information is available, it is likely that, as in animals, elimination of alendronate via the kidney will be reduced in patients with impaired renal function. Therefore, somewhat greater accumulation of alendronate in bone might be expected in patients with impaired renal function (see Section 4.2 Dose and Method of Administration).
5.3 Preclinical Safety Data
Genotoxicity.
Alendronate did not cause gene mutations in bacteria or in mammalian cells in vitro, nor did it cause DNA damage in rat hepatocytes in vitro (alkaline elution assay). In assays of chromosomal damage, alendronate was weakly positive in an in vitro assay using Chinese hamster ovary cells at cytotoxic concentrations (≥ 5 mM) but was negative at intravenous doses up to 25 mg/kg/day (75 mg/m2) in an in vivo assay (chromosomal aberrations in mouse bone marrow).
Carcinogenicity.
No evidence of carcinogenic effect was observed in a 105 week study in rats receiving oral doses up to 3.75 mg/kg/day and in a 92 week study in mice receiving oral doses up to 10 mg/kg/day.6 Pharmaceutical Particulars
6.1 List of Excipients
Microcrystalline cellulose; mannitol; magnesium stearate.
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C in original package.
6.5 Nature and Contents of Container
Alendronate-WGR Once Weekly 70 mg tablets.
PVC/PVDC blister packs of 4 tablets. AUST R 123864.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Alendronate is a white crystalline nonhygroscopic powder. It is soluble in water, very slightly soluble in alcohol and practically insoluble in chloroform.
Bisphosphonates are synthetic analogues of pyrophosphate that bind to the hydroxyapatite found in bone. Alendronate (alendronate sodium trihydrate) is a bisphosphonate that acts as a potent, specific inhibitor of osteoclast mediated bone resorption.
Chemical structure.
 Chemical name: (4-amino-1-hydroxybutylidene) bisphosphonic acid monosodium salt trihydrate.
Chemical name: (4-amino-1-hydroxybutylidene) bisphosphonic acid monosodium salt trihydrate.
Molecular formula: C4H12NNaO7P2.3H2O.
Molecular weight: 325.12.
CAS number.
121268-17-5.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes